Indice del volumen
Volume index
Comité Editorial
Editorial Board
Comité Científico
Scientific Committee
VASCULITIS DE CHURG-STRAUSS:
PRESENTACIÓN CLÍNICA COMO GLOMERULONEFRITIS EXTRACAPILAR NECROTIZANTE PAUCI-INMUNE CON NEFRITIS TUBULO-INTERSTICIAL EOSINOFÍLICA.
Jesús Garrido1, Tânia Sousa1, Fernanda da Cunha2, Edgar Lorga1.
1Unidade de Nefrologia e Diálise do Hospital São Teotónio, Viseu. Portugal.
2Serviço de Anatomia Patológica. Hospitais da Universidade, Coimbra. Portugal.
garrido_nefro @ yahoo.com
Rev Electron Biomed / Electron J Biomed 2004;1:54-62.
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis and periarteritis nodosa. Am J Pathol. 27:277-301, 1951.
- Masi AT, Hunder GG, Lie JT, et al. The American College of Reumathology 1990 Criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 33: 1094-1100, 1990.
- Jennette JC, Falk RJ, Andrassy K et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum. 37:187-192, 1994.
- Watts RA, Lane SE, Bentham G, Scott DGI. Epidemiology of systemic vasculitis. Arthritis Rheum 2000; 43:414-419.
- Reid AJC, Harrison BDW, Watts RA et al. Churg Strauss syndrome in a district hospital. QJM 1998; 91:219-229.
- Murray JF, Nadel JA eds. Textbook of Respiratory Medicine. Philadelphia, Pa: WB Saunders; 1994.
- Martin RM, Wilton LV, Mann RD. Prevalence of Churg-Strauss syndrome, vasculitis, eosinophilia, and associated conditions: retrospective analysis of 58 prescription-event monitoring cohort studies. Pharmacoepidemiol Drug Safety. 1999; 8:179-189.
- Lanham JG, Elkon KB, Pusey CD et al. Systemic Vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine. 1984; 63:65-81.
- Guillevin L, Lhote F, Gayraud M et al. Churg –Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999;78: 26-37.
- Allen JN, Davis WB. Eosinophilic lung diseases. Am J Respir Crit Care Med. 1994; 150:1423-1438.
- Guillevin L, Lhote F, Gayraud M et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996; 75:17-28.
- Tatsis E, Schnabel A, Gross WL. Interferon-alpha treatment of four patients with the Churg-Strauss syndrome. Ann Intern Med 1998; 129:370-374.
- Guillevin L, Cevallos R, Durand-Gasselin B, et al. Treatment of glomerulonephritis in microscopic polyangiitis and Churg-Strauss syndrome. Indications for plasma exchanges, meta-analysis of 2 randomized studies on 140 patients, 32 with glomerulonephritis. Ann Med Interne (Paris) 1997; 148:198-204.
Comentario del Dr. Ernesto Hoffmann. Louisiana State University Medical Center New Orleans, Louisiana. USA
Comentario del Dr. Antonio Félix Conde Martín. Patólogo. Hospital Can Misses. Ibiza. España.
Palabras Clave: Angitis, vasculitis ANCA, glomerulonefritis rápidamente progresiva.
RESUMEN: Se presenta el caso clínico de una mujer de 81 años con antecedentes patología pulmonar obstructiva crónica "idiopática" que desarrolló un cuadro de insuficiencia renal aguda aparentemente prerrenal, con posterior oliguria y eosinofilia simulando una nefropatía túbulo-intersticial alérgica. La evolución atípica y la presencia de p-ANCA, sugerían una vasculitis, patología que se confirmó con la biopsia renal. Los resultados anatomopatológicos revelaron la existencia de una glomerulonefritis necrotizante con semilunas e infiltrado eosinófilo. Estos datos y el historial de la paciente llevaron al diagnóstico de vasculitis de Churg-Strauss. El tratamiento inicial con corticoides y ciclofosfamida y posteriormente con Azatioprina mostró excelentes resultados con mejoría de la función renal, de los parámetros inflamatorios y de la clínica sistémica, que la paciente mantuvo posteriormente.
ABSTRACT: We report a case of a 81 year-old woman with idiopathic chronic obstructive pulmonary disease, who developed a functional acute renal failure with delayed oliguria and eosinophily, simulating an acute interstitial nephropathy. The unusual clinical course and the presence of antimyeloperoxidase antibodies (p-ANCA) suggested a vasculitis; the renal biopsy confirmed this diagnosis. The histology revealed a crescentic glomerulonephritis with eosinophilic infiltration. With these data and the pulmonary history of the patient, a diagnosis of Churg-Struss Vasculitis was made. The initial treatment with steroids and ciclophosphamide switched later to azatioprine, was succeeded with a sustained improvement in renal function, inflammatory markers and clinical course.
INTRODUCCIÓN
Las vasculitis continúan siendo una patología difícil, tanto por la afectación multisistémica como por la diferente forma de presentación y evolución clínica. El síndrome de Churg-Strauss fue descrito por Jacob Churg y Lotte Strauss en 1951 como una vasculitis sistémica eosinofílica1. En 1990 el Colegio Americano de Reumatología definió los criterios de diagnóstico2 y en 1994, en la Conferencia de Chapel-Hill3, se describió como "asma y eosinofilia asociadas a granulomas inflamatorios con afectación del tracto respiratorio con vasculitis necrotizante de pequeños y medianos vasos".
De etiología desconocida, su incidencia se estima en 2-4 casos por millón de habitantes en la población general y entre 12.5-20 veces mas, hasta 64 casos por millón en los asmáticos 4,5,6,7. Se manifiesta con igual frecuencia en ambos sexos, en tres fases8,9;
- I) Inicial: entre los 20-40 años con asma y rinitis alérgica.
II) Eosinofilia periférica e infiltración de órganos.
III) Vasculitis, con afectación sistémica: Pulmonar (asma grave, tos, disnea, sinusitis, hemorragia...), neurológica 78% (mononeuritis múltiple, polineuropatia), cutánea 50% (púrpura, lívedo reticularis, urticaria), cardiovascular (pericarditis, IAM), gastrointestinal (gastroenteritis eosinofílica, dolor, diarrea, hemorragia).
Hasta un 25% de los casos presentan afectación renal con nefropatia túbulo intersticial (NTI) frecuente y glomerulonefritis focal y segmentar o necrotizante con semilunas. No obstante el tratamiento con esteroides e inmunosupresores ha mejorado drásticamente la evolución y el pronóstico de esta patología.
CASO CLÍNICO
Mujer de 81 años con antecedentes de diabetes mellitus tipo 2, hipertensión arterial e enfermedad pulmonar obstructiva crónica de etiología desconocida con oxigenoterapia domiciliaria. Sin antecedentes de insuficiencia renal conocida.
Acudió al Servicio de Urgencias por diarrea, fiebre y molestias urinarias, observándose alteración importante de la función renal, interpretada como un fracaso renal agudo prerrenal. Inició antibioterapia con una cefalosporina de 3ª generación y reposición hidro-electrolítica con evolución inicial favorable. Las hemoculturas fueron positivas para staphilococcus hominis sensible a ciprofloxacino, antibiótico que inició. Posteriormente presentó un cuadro de oliguria, fiebre y eosinofilia junto con deterioro de la función renal, interpretado como una nefritis túbulo-intersticial alérgica (por furosemida vs antibiótico), necesitando hemodiálisis durante una semana.
Suspendió dichos fármacos y comenzó corticoterapia oral (día 12º), con desaparición de la fiebre, normalización de la eosinofilia, recuperación de la diuresis y ligera disminución de la creatinina plasmática (Crp), destacando un nuevo aumento de los eosinófilos y de la Crp de 3.5 mg/dl a 5.7 mg/dl, paralelo a la disminución de los corticoides.
Las pruebas complementarias revelaron, además de un discreto infiltrado pulmonar bilateral, la presencia de anticuerpos anticitoplasma de neutrófilo antimieloperoxidasa (p-ANCA). Se realizó una biopsia renal percutánea y ese día inició tratamiento empírico con corticoides (3 bolos de 500 mg de 6-metilprednisolona y después oral 1 mg/kg/dia). Los resultados anatomopatológicos (tabla 1 y figuras 1-7) mostraron una glomerulonefritis necrotizante proliferativa extracapilar, con granulomas y nefritis tubulointersticial con infiltrado de eosinófilos. Con el diagnóstico de vasculitis de Churg-Strauss, se asoció ciclofosfamida oral (1.5 mg/kg/día) con evolución favorable de la función renal y de la patología respiratoria. Tuvo alta con 3.5 mg/dl de creatinina plasmática.
|
Estudio Microscópico : cilindro de parénquima renal de 18 mm de longitud con 9 glomérulos, de los cuales 2 presentan ligera expansión mesangial. De los restantes, 3 están completamente esclerosados y 4 presentan proliferación extracapilar con lesiones importantes activas de necrosis celular, reconociéndose la presencia de granulomas glomerulares que rompen la cápsula y destruyen la totalidad del glomérulo, donde solo se reconocen algunos restos de la membrana basal. Los glomérulos están transformados en granulomas centralmente hialinos o necróticos con fibrina y un extenso proceso inflamatorio que rompe la cápsula y se extiende al intersticio. En uno de los glomérulos la proliferación extracapilar es mas segmentar aunque produce colapso parcial del ovillo capilar. Hay depósitos difusos y segmentares de fibrina formando trombos intracapilares. Aparece también nefritis intersticial aguda importante con un infiltrado mixto donde predominan los linfocitos y plasmocitos, teniendo asimismo neutrófilos y eosinófilos. Este infiltrado celular forma agregados densos inflamatorios. Hay lesión tubular importante en las zonas de mayor infiltrado intersticial que atraviesa la pared, con células inflamatorias en la luz tubular totalmente obliterada y necrosis celular con cilindros de material granular eosinófilo en el interior de muchos túbulos, particularmente en los distales. Las arteriolas y arterias interlobares presentan espesamiento concéntrico de la pared, sin depósitos de fibrina reconocibles pero con prominencia de las células endoteliales; Están envueltas íntimamente por el proceso inflamatorio intersticial con disposición periadventicial (periarteritis). Figuras 1-7. |
|
Estudio de Inmunofluorescencia: incluye 5 glomérulos de los cuales 2 están hialinizados, presentando el resto lesiones de proliferación y esclerosis. Solo se observan depósitos glomerulares mínimos de IgM y C3 en pequeños segmentos que corresponden con sinequias del ovillo capilar a la cápsula en evolución a esclerosis. Hay también depósitos de fibrinogeno (+) que dibujan el endotelio de los capilares glomerulares y de las pequeñas arterias.
|
|
Diagnóstico Histopatológico: GLOMERULONEFRITIS PROLIFERATIVA EXTRACAPILAR, NECROTIZANTE CON SEMILUNAS, "PAUCI-INMUNE", COMPATIBLE CON PROCESO DE VASCULITIS SISTÉMICA.
|
Tabla 1. Resultados histopatológicos de la biopsia renal.
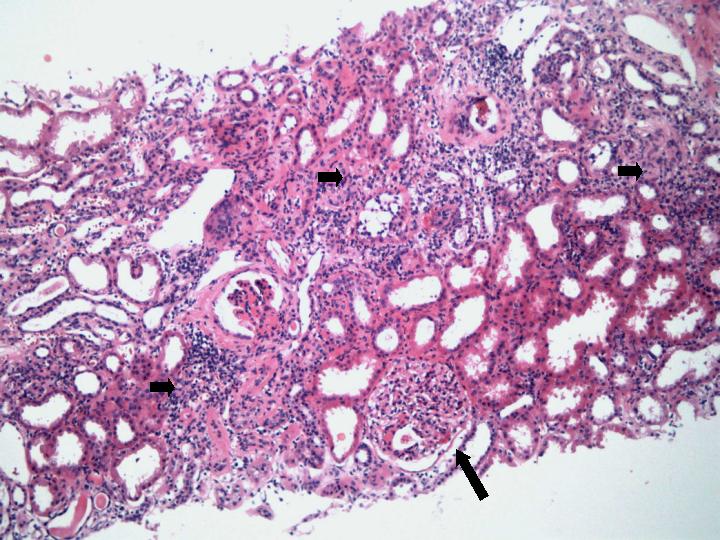
Fig 1. Glomerulonefritis proliferativa y necrotizante (flecha larga) con nefropatia intersticial (Flechas cortas) (H-E x 100).
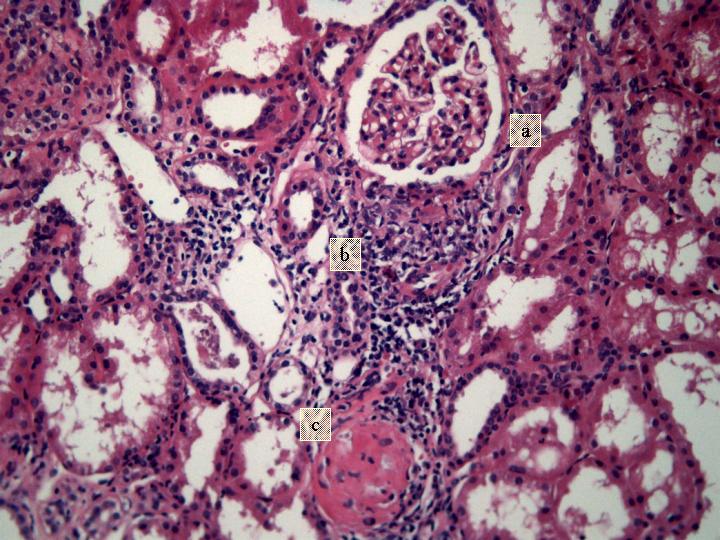
Fig 2. Glomérulo normal (a). Gramuloma con destrucción glomerular (b). Glomérulo hialino (c) (H-E x 200).
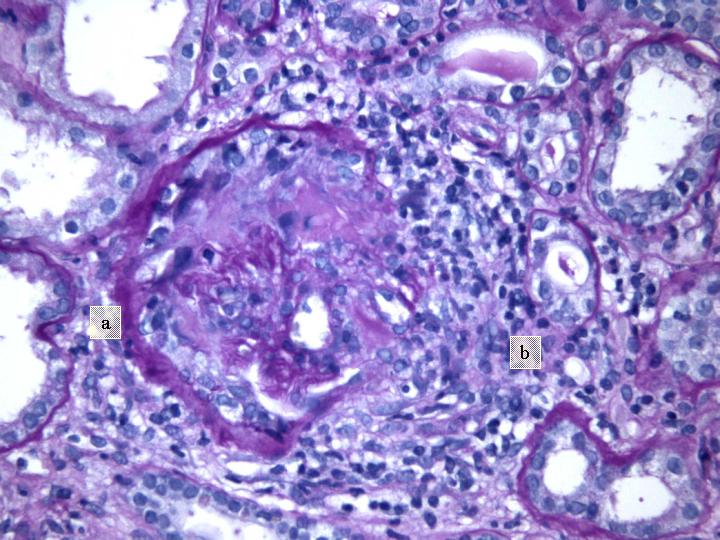
Fig 3. Glomérulo con proliferación extracapilar y necrosis (a). Reacción inflamatoria periglomerular (b) (PAS x 200).
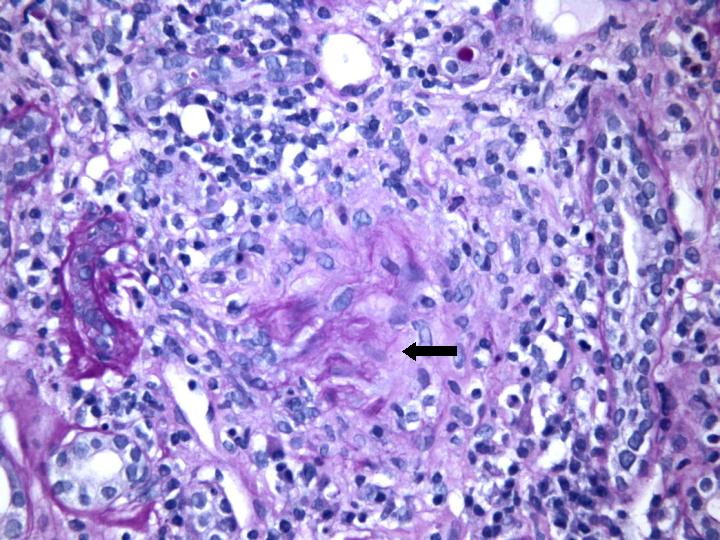
Fig 4. Proceso necrotizante glomerular con restos de mesangio y membrana basal (PAS x 400).
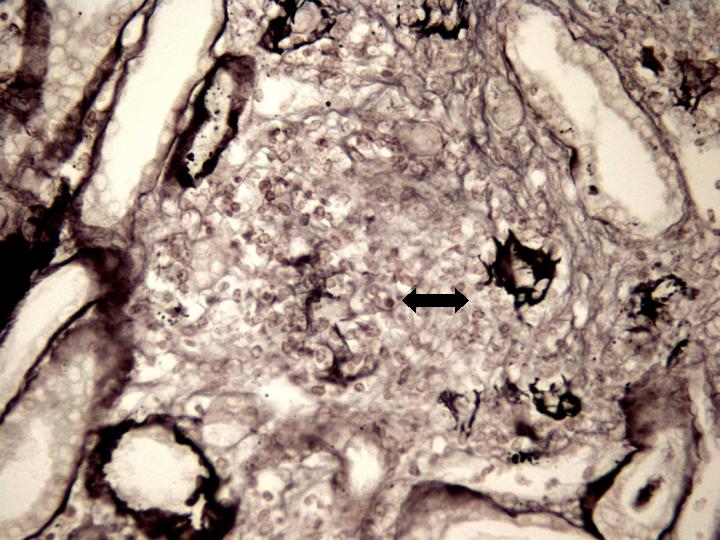
Fig 5. Glomérulo envuelto por el proceso granulomatoso y necrotizante con restos de la membrana basal glomerular (Marinozi x 400).
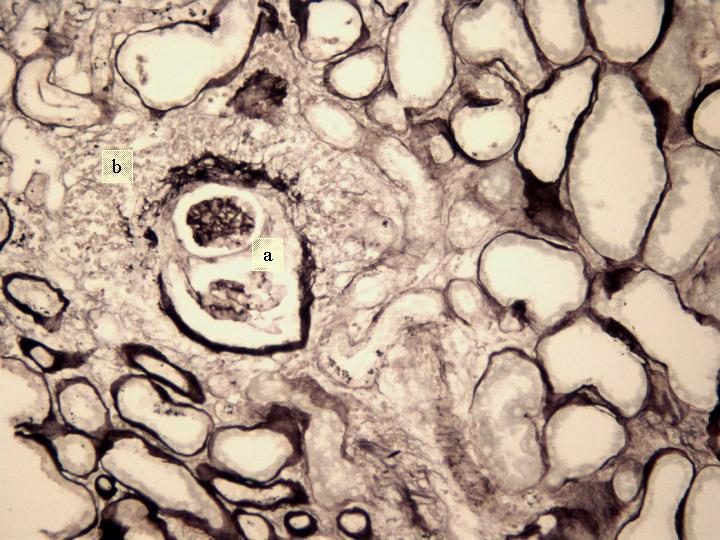
Fig 6. Glomérulo con esclerosis segmentar secundaria (a) y granuloma periglomerular (b). (Marinozi x 400).
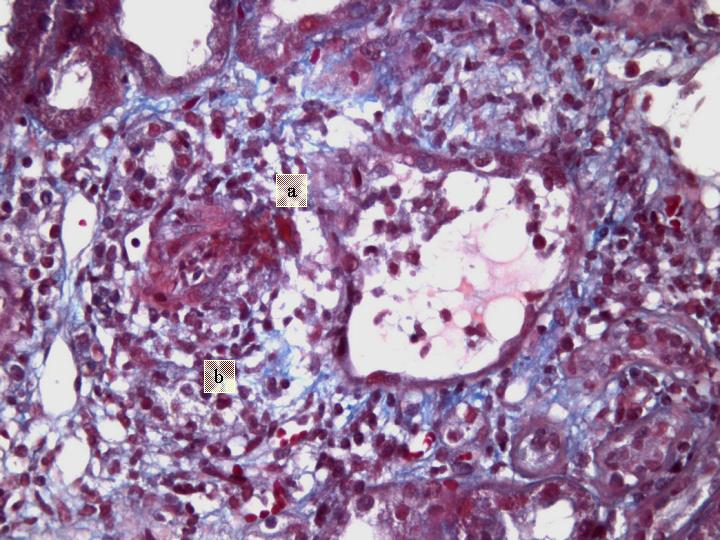
Fig 7. Glomérulo con necrosis y depósitos de fibrina (a) y granuloma envolvente (b) (Tricrómico de Masson x 200).
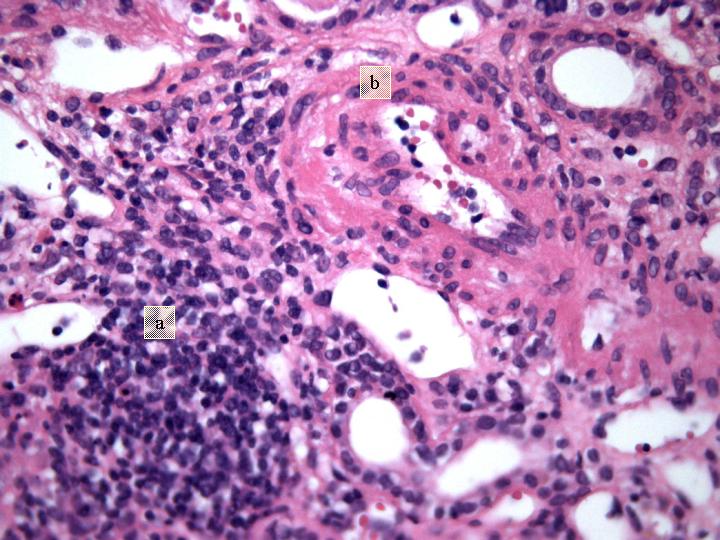
Fig 8. Nefritis intersticial con lesión tubular (a) y vascular (b) (H-E x 400).
Durante el seguimiento posterior continuó con una disminución progresiva de la creatinina y suspendió la oxigenoterapia domiciliaria. Mantuvo los corticoides en dosis decreciente hasta su suspensión en el 7º mes. Debido a la leucopenia secundaria a la CFM a pesar de la reducción de la dosis, fue substituida por azatioprina (1,5mg/kg/día v.o.) en el tercer mes. La paciente tuvo una mejoría clínica significativa y una evolución favorable de los parámetros analíticos (normalización de la VSG, disminución de la Crp y de los p-ANCA, estabilidad de los leucocitos sin eosinofilia y aumento e la hemoglobina sin necesidad de eritropoyetina). Un año después de iniciar el tratamiento inmunosupresor, la paciente se encuentra clínicamente bien, con recuperación importante de la función renal (tablas 2-3) y asintomática a nivel respiratorio.
Tabla 2. Parámetros analíticos durante el primer año de tratamiento.
|
Mes 1 |
Mes 3 |
Mes 6 |
Mes 12 |
|
|
Creatinina plasma mg/dl |
2.8 |
2.3 |
1.9 |
1.7 |
|
Urea plasma mg/dl |
127 |
100 |
73 |
68 |
|
CCr ml/min |
20 |
26 |
30 |
38 |
|
Proteinuria g/24 h |
1.01 |
- |
0.74 |
0.52 |
|
Sedimento urinario |
Leucos +
|
- |
Leucos + |
Leucos - |
|
Hemoglobina g/dl |
11.2 |
12.1 |
14.6 |
14.8 |
|
Leucocitos sangre perif |
4400 |
1900 |
6700 |
6200 |
|
Eosinófilos sangre perif |
1% |
7% |
1.9% |
2% |
|
p-ANCA |
82 |
49 |
- |
50 |
DISCUSIÓN
Destaca en ese caso clínico, la forma atípica de presentación con diarrea y fiebre simulando un cuadro de IRA prerrenal que mejoró con la introducción de los corticoides ante lo que parecía ser una NTI alérgica. La evolución favorable del cuadro inicial con los esteroides, reforzaría posteriormente que toda la clínica inicial podría formar parte de una misma entidad: la activación de una vasculitis sistémica con afectación pulmonar (infiltrado intersticial), digestivo (se ha descrito una diarrea eosinofílica asociada a este síndrome, aunque no se realizó biopsia intestinal) y renal. La revisión del historial clínico de la paciente reveló datos fundamentales: sinusitis, eosinofilia crónica desde 6 años antes, patología pulmonar crónica grave con espirometría característica con patrón obstructivo, aumento del volumen residual y disminución de la difusión de CO. Estos síntomas y signos junto con la positividad de los p-ANCA (hasta en dos tercios de los pacientes) sugerían de hecho una vasculitis de Churg-Strauss, diagnóstico que se confirmaría con los resultados de la biopsia. Además de la glomerulonefritis necrotizante extracapilar, la histología mostró la presencia de granulomas (poco frecuentes) y un característico infiltrado intersticial por eosinófilos.
El diagnóstico con 4 de los 6 criterios del ACR (asma, eosinofilia periférica > 10%, mono o polineuropatia, infiltrado pulmonar, alteraciones de los senos paranasales, biopsia con acúmulos de eosinófilos en áreas extravasculares), tiene una sensibilidad y una especificidad del 88.5 y 99.7% respectivamente2. El diagnóstico diferencial de la angitis de Churg-Strauss debe incluir, otras vasculitis, la neumonía eosinofílica, la aspergilosis pulmonar alérgica, reacciones medicamentosas o infecciones, entre otros. Además del Churg-Strauss, son varias las vasculitis de pequeño vaso que asocian la presencia de ANCA con la afectación renal y pulmonar como la granulomatosis de Wegener (con predominio de los ANCA tipo antiproteinasa, "C-ANCA") y la poliangeitis microscópica. Se ha descrito tambien en la enfermedad de Goodpasture la presencia de ANCA asociados a los anticuerpos antimembrana basal. La presentación clínica de estas últimas, suele ser mas agresiva habitualmente con afectación pulmonar hemorrágica. A nivel renal generalmente se presentan como una glomerulonefritis extracapilar necrotizante con insuficiencia renal rapidamente progresiva y su tratamiento de forma general no depende del tipo etiológico sinó de la gravedad clínica (habitulamente con corticoides, inmunosupresores y/o plasmaféresis). La presentación de la vasculitis de Churg-Strauss suele ser mas leve y progresiva y la afectación renal fundamentalmente intersticial aunque están descritos casos de glomerulonefritis. En el caso que presentamos, además de la clínica típica respiratoria y la afectación tubulo-intersticial destaca la agresividad de la vasculitis a nivel glomerular, similar a la mostrada por la vasculitis de Wegener o la poliangeitis microscópica.
El tratamiento fundamentalmente con esteroides (0.5-1 mg/Kg con disminución progresiva en 3-6 meses) ha mejorado drásticamente el curso y el pronóstico de la vasculitis de Churg-Strauss. La remisión clínica alcanza hasta el 90% de los casos tratados, de los cuales un 25% presentan recidivas, generalmente asociadas a una disminución en la dosis de corticoides y acompañada de un aumento de la eosinofilia 9.
Se estima que sin corticoides la mortalidad supera el 50% a los 3 meses del diagnóstico, con supervivencia superior al 70% a los 6 años en los tratados 9,10; La afectación cardiaca, gastrointestinal, neurológica central o renal (Crp > 1.6 mg/dl, proteinuria > 1 gr/día) se asocia a peor pronóstico11. En estos casos está indicado el uso de corticoides a altas dosis (1.5 mg/kg durante largos periodos) y la asociación de inmunosupresores. La ciclofosfamida, la azatioprina y la Inmunoglobulina endovenosa ofrecen buenos resultados en los casos de enfermedad grave, fulminante o resistencia a los esteroides. El uso de CFM está indicado en la fase aguda hasta conseguir la remisión o el control de la enfermedad, siendo habitual la substitución por AZA como tratamiento de mantenimiento, dado que presenta menos efectos secundarios y complicaciones. Se ha descrito beneficio con la combinación de corticoides e Interferón-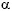 en los casos de resistencia a la ciclofosfamida12. La plasmaféresis no ha demostrado beneficios superiores 9,13.
en los casos de resistencia a la ciclofosfamida12. La plasmaféresis no ha demostrado beneficios superiores 9,13.
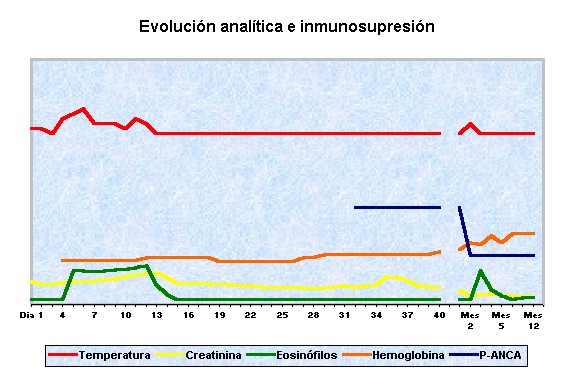
Tabla 3. Gráfico de las alteraciones analíticas relacionadas con la inmunosupresión.
CONCLUSIONES
Las vasculitis continúan siendo una patología compleja tanto por la variedad en la forma de presentación como por la afectación sistémica. El diagnóstico del síndrome de Churg-Strauss requiere además de la sospecha clínica un diagnóstico definitivo basado en el estudio histo-patológico. El tratamiento agresivo con corticoides y ciclofosfamida incluso en pacientes de edad avanzada con patologia grave, puede ser determinante para alcanzar una mejoría clínica significativa e mejorar el pronóstico.
BIBLIOGRAFIA
___________
Correspondencia:
Jesús Garrido García.
Unidade de Nefrologia e Diálise. Hospital São Teotónio.
Av. Rei D. Duarte, 3504-509, Viseu, Portugal.
e-mail: garrido_nefro@yahoo.com
Comentario del Dr. Ernesto Hoffmann. Louisiana State University Medical Center New Orleans, Louisiana. USA
La presentación de un caso raro renueva mi interés por un tema tan confuso como son las vasculitis. Creo que el presente caso esta bien documentado y esta dentro del limite para su aceptación como un caso de Vasculitis de Churg-Strauss. La presentación esta bien organizada y con referencias adecuadas.
Estoy seguro que su publicación despertara el interés de los lectores por este tema.
Comentario del Dr. Antonio Félix Conde Martín. Patólogo. Hospital Can Misses. Ibiza. España.
La afectación gastrointestinal clínica y patológica es un hecho frecuente en el contexto del Síndrome de Churg-Strauss. Sin embargo es infrecuente el debut de la enfermedad como cuadro digestivo. Los autores presentan un caso que se diagnostica tras cuadro de diarrea e insuficiencia renal aguda, inicialmente interpretada como pre-renal. La observación de la evolución del caso, la mejoría tras instaurar tratamiento corticoideo y la revisión de los antecedentes clínicos llevan al diagnóstico correcto.
Se realiza una detallada descripción de la evolución clínica en relación al tratamiento y de los hallazgos patológicos de la biopsia renal.
Recibido: 28 de Enero de 2004.
Publicado: 19 de Febrero de 2004