Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
PORFIRIA CUTÁNEA TARDÍA
Ornella Agnese, Rocio Macarena Sueiro-Lopez, Iara Yamila Taito-Vincenti, Sabrina Bassi
Instituto Universitario del Hospital Italiano de Buenos Aires,
Argentina
Email: ornella.agnese @ hospitalitaliano.org.ar
Rev Electron Biomed / Electron J Biomed 2019;2:53-61.
Comentario de la revisora Dra. Paula A. Enz MD. Dermatóloga y Geriatra. Sub-Jefa del Servicio de Dermatología del Hospital Italiano de Buenos Aires, Argentina.
Comentario de la revisora Dra. Anita Amalia Rossi MD Dermatóloga consultora del Hospital de Nińos de La Matanza. Provincia de Buenos Aires, Argentina.
RESUMEN
Las porfirias son un grupo de enfermedades metabólicas raras generadas por bloqueos en la vía de síntesis del hemo que generan aumento de la excreción de porfirinas o sus precursores. La porfiria cutánea tardía se genera por una falla en la enzima "Uroporfirinógeno decarboxilasa", que generaría de decarboxilación del uroporfirinógeno III en Coproporfirinógeno III.
Esta porfiria puede dividirse en tres tipos: Tipo I o esporádico, que se manifiesta con un nivel de enzima del 50% en el hígado; Tipo II o familiar que se caracteriza por una deficiencia del 50% en la actividad de la misma enzima en varios tejidos como hígado y eritrocitos; y el Tipo III o porfiria hepatoeritropoyética.
El diagnóstico de la misma se basa en los antecedentes clínicos y familiares, el examen físico, y análisis de laboratorio como espectrofotometría, una biopsia hepática, y el análisis del Gen URO-D para buscar mutaciones que condicionen su malfuncionamiento. Este análisis se realiza a través de amplificación por PCR y utilizando métodos de secuenciación como Illumina, SOLiD, Ion Torrent y PacBio.
El tratamiento de la porfiria cutánea tardía es sintomático para las lesiones cutáneas y reduciendo los niveles de hierro circulante utilizando flebotomías o medicación. Posteriormente teorizamos como podría llegar a aplicarse una terapia génica en esta enfermedad.
PALABRAS CLAVE: porfiria cutánea tardía, diagnóstico, tratamiento
ABSTRACT: PORPHYRIA CUTANEA TARDA
Porphyrias are a group of rare metabolic diseases generated by blockages in the heme synthesis pathway that lead to increased excretion of porphyrins or their precursors. The porphyria cutanea tarda is generated by a failure in the enzyme "uroporphyrinogen decarboxylase", which would generate decarboxylation of uroporphyrinogen III in coproporphyrinogen III.
This porphyria can be divided into three types: sporadic (type I), which manifests itself with an enzyme level of 50% in the liver; family (type II) that is characterized by a 50% deficiency in the activity of the same enzyme in various tissues such as liver and erythrocytes; and hepatoerythropoietic porphyria (type III).
Its diagnosis is based on the clinical and family history, the physical examination, and laboratory analysis such as spectrophotometry, liver biopsy, and URO-D Gene analysis to look for mutations which condition its malfunction. This analysis is performed through PCR amplification and using sequencing methods such as Illumina, SOLiD, Ion Torrent and PacBio.
Treatment of porphyria cutanea tarda is symptomatic for skin lesions and by reducing circulating iron levels using phlebotomies or medication. Later we theorize how gene therapy could be applied in this disease.
KEY WORDS: porphyria cutanea tarda, diagnosis, treatment
INTRODUCCIÓN
Las porfirias son un grupo de enfermedades metabólicas raras caracterizadas por aumento de la excreción de porfirinas o sus precursores, que se genera por bloqueos en la vía de síntesis del hemo. Las mismas pueden ser hereditarias o adquiridas, afectando a personas en distintos rangos etarios dependiendo del tipo específico de porfiria 1,2.
con las enzimas participantes y el tipo de porfiria generada si estas se ven afectadas.
ALA: ácido deltaaminolevulínico; PBG: porfobilinógeno.
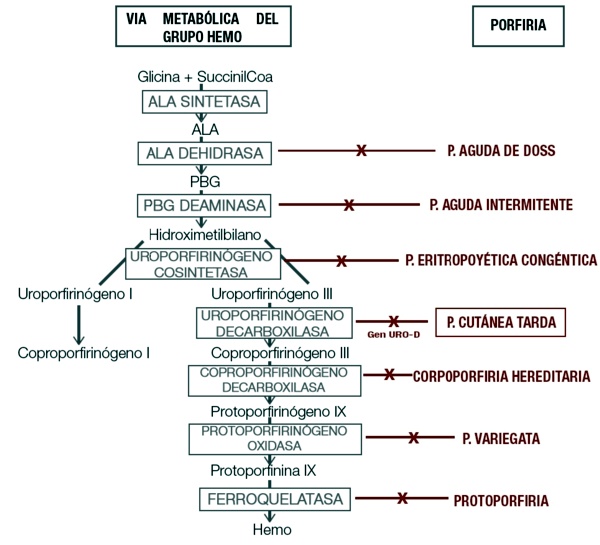
Podemos clasificar las porfirias en: agudas, cutáneas o mixtas3. También pueden ser clasificadas según el sitio principal de acumulación de los intermediarios del hemo, en: hepáticas, eritropoyéticas y hemato-eritropoyéticas1,4.
Las porfirias no son prevalentes, por esto son consideradas como enfermedades raras. Su prevalencia varía entre 0,5 a 10 por 100.000 personas en diferentes poblaciones (presenta una incidencia de 1:5000 en República Checa y Eslovaquia pero en Estados unidos es de 1:25000) 1,5,6. La porfiria más frecuente en argentina es la Porfiria Aguda Intermitente (PAI) con una prevalencia de 1:100.000, seguida de la Porfiria Variegata, con una frecuencia de 1:600.000. Sin embargo, la porfiria más frecuente en la Argentina es la Porfiria Cutánea Tardía, con una prevalencia de 1:37.000. 3,7.
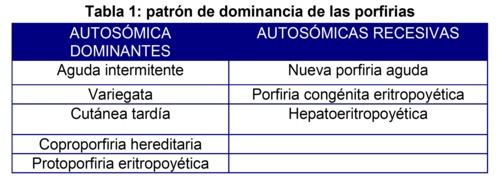
Todas las porfirias tienen origen en desórdenes genéticos, a excepción de la porfiria cutánea tipo I, aunque varían en su dominancia, pudiendo ser autosómicas dominantes o recesivas (Tabla 1).
En este trabajo se desarrollará la Porfiria Cutánea tardía, la cual se debe a la deficiencia parcial, tanto hepática como eritrocitaria, de la enzima uroporfirinógeno III descarboxilasa. La misma participa como la quinta enzima del proceso de síntesis del hem, encargada de la remoción de átomos de oxígeno y carbono del intermediario uroporfirinógeno III para formar coproporfirinógeno III8. Se profundizará más sobre los tipo II y III ya que son aquellos en los que existe un componente genético en el desarrollo de la enfermedad.
PORFIRIA CUTÁNEA TARDÍA
En 1978 se clasificó la porfiria cutánea tardía en dos tipos. El tipo I o esporádico, que se manifiesta con un nivel de enzima del 50% en el hígado, y el tipo II o familiar que se caracteriza por una deficiencia del 50% en la actividad de la misma enzima en varios tejidos como hígado y eritrocitos, generado por un defecto genético6. Actualmente también se hace referencia a la porfiria hepatoeritropoyética como porfiria cutánea tardía tipo III.
TIPO II O FAMILIAR
El tipo II o familiar solo representa el 20% del total de los casos de porfiria cutánea tarda. La causa de este tipo es la presencia, en heterocigosis, de una alteración en el gen URO-D, ubicado en el brazo corto del cromosoma 1, en la posición 34.1 (1p34.1), el cual posee 10 exones en un total de 3kb 9 y codifica para una proteína de 367 aminoácidos y una masa molecular de 40.8 kD9. Dicha proteína es la enzima uroporfirinógeno descarboxilasa, que genera la conversión uroporfirinógeno a coproporfirinógeno a través de la eliminación de las cuatro cadenas laterales de carboximetilo10.
Se han identificado 50 mutaciones diferentes asociadas a esta enfermedad y también factores ambientales desencadenantes13. El ARNm mensajero es el la variante del transcripto número 2 (identificada como NR_036510.1)14, y a partir del mismo se indican los en la proteína. En Argentina, la mutación más hallada fue H63D, que produce una mutación missense de Histidina por Aspartato en la posición 63 de la proteína. Esto significa que los codones CAU, CAC que codifican para histidina, cambian su primera base de una citocina a una guanina, para dar los codones codificantes para aspartato GAU, GAC15.
Por otro lado presenta una penetrancia incompleta3, por lo que solo un 10% de los portadores de esta alteración manifiesta la enfermedad8. Dicha expresión fenotípica puede verse influenciada por otros factores ambientales, entre los que podemos mencionar el uso de alcohol y tabaco, exceso de hierro, exposición a químicos aromáticos polihalogenados, estrógenos, hepatitis, infecciones por VIH y hemocromatosis hereditaria por mutaciones en el gen HFE 5,6,10,14. Estos desencadenan la enfermedad ya que reducen la actividad de la enzima uroporfirinógeno III descarboxilasa del 50% al 20%, y generan así la acumulación de porfirinas10.
En Argentina, la porfiria cutánea tardía del tipo I es más frecuente en hombres (4:1), probablemente debido a la mayor exposición a factores desencadenantes, siendo el virus de la hepatitis C el más prevalente (35,2 %)15. Por otro lado, la variante tipo II se cree que afecta a hombres y mujeres en igual proporción8.
En la porfiria cutánea tardía el principal síntoma es la fotosensibilidad cutánea, manifestándose en ampollas y lesiones de la piel ante la exposición a la luz1. Otro signo de la enfermedad es la coloración rojiza o café de la orina, generada por la eliminación de las porfirinas a través de la misma1,6. Este tipo de porfiria no posee manifestaciones neuro viscerales6.
TIPO III O HEPATOERITROPOYÉTICA
Anteriormente se mencionó que si la mutación en el gen URO-D se encuentra en heterocigosis se desarrolla PCT tipo II, pero también existe la posibilidad que la alteración se presente en ambos alelos (homocigosis). Este es el caso de la denominada porfiria cutánea tardía tipo III o porfiria hepatoeritropoyética. En estos pacientes la actividad enzimática es extremadamente baja (7-8%), tanto en el tejido hepático como en los eritrocitos, por lo que se manifiesta en los primeros ańos de vida y de manera más grave que el tipo II5,6,10. En Argentina solamente se han identificado alrededor de 30 casos de este tipo5.
DIAGNÓSTICO
El diagnóstico en general puede establecerse al considerar los antecedentes clínicos y familiares, el examen físico, y análisis de laboratorio apropiados, como espectrofotometría o una biopsia hepática6,15.
El diagnóstico de la porfiria cutánea tardía de tipo familiar se establece en el caso índice mediante los indicadores nombrados anteriormente y mediante la identificación de una variante patogénica en heterocigosis en el gen URO-D. Para esto se utilizan distintas técnicas de diagnóstico genético, las cuales pueden ser de un solo gen o de un panel multi genético.
Primero se amplifica en el gen por medio de la técnica de PCR y luego se procede al análisis del mismo mediante secuenciación, preferencialmente Next generation sequencing como se explicará en el próximo párrafo. Si ninguna variante patológica fue encontrada se procede a un análisis de deleción o duplicación en el gen, en el cual se pueden utilizar varios métodos como PCR cuantitativa, PCR de largo rango (que permite la amplificación de longitudes de ADN que generalmente no se pueden amplificar por métodos de PCR rutinarios)16, MLPA ("multiplex ligand probe amplification", que suele utilizarse para amplificar simultáneamente los exones de un gen utilizando primers para cada uno) o microarray17. En la actualidad, debido a los bajos costos, se está reemplazando la secuenciación clásica por la tecnica de next generation sequencing18. Estas técnicas incluirían Illumina, SOLiD, Ion Torrent y PacBio.
En el caso de la detección del Gen URO-D, existe un estudio (GTR000519417.9) que usando Next generation Sequencing (NGS) cubre la región codificante del gen, incluyendo 10 bases de ADN no codificante flanqueando cada exón que se amplificará. El ADN se extrae de la muestra y se secuencia utilizando el método Illumina. Esta técnica utiliza oligonucleótidos unidos a una matriz y terminadores-marcados de unión reversible. Los fragmentos se unen a los oligonucleótidos y son ampliados, pero los terminadores que se remueven luego de cada ciclo para reutilizarse. Los resultados se comparan a través de programas computacionales, y las variantes patogénicas (clase V), probablemente patogénicas (clase IV) y de significado incierto (variante III) se confirma utilizando secuenciación Sanger. Se analizan además polimorfismos de repeticiones en tándem ya que ciertos podrían relacionarse con la funcionalidad de la enzima, estos se confirman con aCGH, MLPA, o PCR antes de ser reportados19.
Dentro del panel multigenético no solo se incluye el gen UROD sino también otros de interés que pueden aportar a un diagnóstico diferencial. Este panel podría ser el "Panel de secuenciación para porfirias cutáneas/crónicas con detección de CNV (variación del número de copias)" (dicho código de pedido es GTR000552988.4), que incluye además de la detección de la porfiria cutánea tardía del tipo II (del Gen UROD del cromosoma 1p34.1), la protoporfiria eritropoyética ligada al X (gen ALAS 2 del cromosoma Xp11.21); la porfiria eritropoyética congénita (gen UROS del cromosoma 10q26.2); la protoporfiria eritropoyética (gen FECH del cromosoma 18q21.31); la Coproporfiria hereditaria (gen CPOC del cromosoma 3q11.2) y la porfiria variegata (gen PPOX del cromosoma 1q23.3)20.
Por su carácter de penetrancia incompleta, el diagnóstico presintomático en las familias afectadas es fundamental para la detección de los portadores asintomáticos y su asesoramiento para evitar la exposición a compuestos porfirinogénicos a fin de prevenir el desencadenamiento de la enfermedad3.
En argentina existe un centro dedicado exclusivamente al diagnóstico tanto genético como bioquímico de todas las porfirias. Es el Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP) dependiente del Hospital de Clínicas y del CONICET21.
TRATAMIENTO
Actualmente el tratamiento de la porfiria cutánea tardía es sintomático para las lesiones cutáneas14. Además, se busca la reducción de los niveles de hierro circulante a través de flebotomías 22,23 o del tratamiento con medicación, como la cloroquina y la deferoxamine. La fotoprotección tanto física con ropa como en la forma tópica con protectores que tengan filtros UVB y UVA de alta protección son parte importante de estos tratamientos.
La cloroquina bloquea las porfirinas y ayuda a su excreción, reduciendo así sus niveles en el organismo. Se debe indicar con sumo cuidado ya que incluso en las dosis normales puede generar necrosis hepática aguda24. Sin embargo, la respuesta al tratamiento de cloroquina depende del perfil genético del paciente: aquellos que tienen un polimorfismo C282Y heterocigota en el gen HFE, (codificante para la proteína homónima encargada de la regulación de la absorción del hierro) responden a este medicamento; mientras que si se es homocigota padece de hemocromatosis hereditaria, y no responde2. Otra alternativa para el tratamiento es la deferoxamina, que es un agente quelante que promueve la remoción del exceso de hierro. Del mismo se realizó un estudio piloto en fase 3 (código identificador: NCT00599326) como tratamiento para la porfiria cutánea tardía25.
Propuesta de terapia génica
Al deducir que los desencadenantes de la expresión de esta enfermedad son la sobrecarga de la vía metabólica (por exceso de hierro) y la disminución de actividad de esta enzima (por ejemplo, por lesión hepática durante hepatitis); suponemos que un método posible para la mejoría de esta condición es la terapia génica de adhesión de un gen nuevo funcional, o bien la reparación del alelo mutado.
Para la transfección del nuevo material genético dispondremos de vectores virales, sin embargo el más utilizado es el Virus Adeno-Asociados recombinante debido a su excelente perfíl de seguridad en comparación al Adeno-Virus. Además al incorporarse al torrente sanguíneo el primer sitio de transfección es el hígado26.
Adenovirus (AV) Virus Adeno-Asociados (AAV)
Transfecta células en división y células quiescentes.
Se deposita episomalmente, no se integra al genoma. Mayoritariamente se deposita episomal. Puede integrarse al genoma (preferencias por el brazo q13.4-ter del cromosoma 19)
Generan una respuesta inmune. Activa linfocitos citotóxicos, linfocitos CD4+; que destruyen las células transfectadas. Activa linfocitos B, cuyos anticuerpos dificultan una consiguiente administración de más virus. Respuesta inmune menor. Puede erradicarse al inhibir la expresión del toll-receptor 9.

Si bien el virus adeno-asociado es el más seguro, también presenta ciertas desventajas.
Para su obtención requiere una co-transfección con dos plásmidos un plásmido helper que contenga el transgen flanqueado por repeticiones terminales invertidas (ITRs) y otro que codifique para genes de replicación, de cápsula y proteínas adenovirales (ya que requiere de una "infección conjunta" con adenovirus)28.
El serotipo a utilizar es controversial. En modelos murinos el serotipo 8 logró un 40% de transfección, y mostró una eficacia mayor que los serotipos 2 y 526,27. Sin embargo se cree que en los humanos sería más eficiente una cápside quimera que transporte 5 cápsides parentales de Virus adeno-asociados diferentes28.
Una vez discutido el vector, dentro de su genoma podríamos incluir una copia del gen URO-D. Este podría recombinarse homólogamente con el alelo mutado de la célula transfectada y repararlo28, o mantenerse episomal y ser transcripto para aumentar la cantidad de enzima funcional. Otro enfoque para la reparación del ADN podría ser integrar un método para reparar la mutación puntual que posee. Podría el mismo ser incorporado dentro de la célula usando un vector viral.
Actualmente está en desarrollo una nueva estrategia experimental para corregir enfermedades metabólicas generadas por la mutación de un solo nucleótido, que se basa en un quimeróplasto27. Los quimeroplastos son oligonucleótidos que combinan ADN y ARN (en la zona de oligonucleótidos target), metilados para resistir las endonucleasas. Estos poseen mayor actividad de recombinación y están diseńados para hibridar con el ADN target en el genoma. Una vez unido, induce un cambio missmatch y se utiliza el quimeroplasto como molde para la reparación de la mutación puntual28.
Utilizando como ejemplo la mutación más común en Argentina (H63D) podría crearse un quimeroplásto que indique un missmatch en la primera base del codón que codifica para la posición 63, que sería una guanina, para que al repararse volviese a incluirse una citosina y se recupere el aminoácido histidina. Siguiendo este ejemplo, podrían corregirse otras mutaciones de este gen que se deban a un cambio de base que genere una mutación missense o una mutación puntual que modifique el sitio de splicing de un intrón.
Por último, el sistema CRISP/Cas9 podría servir al momento de corregir una mutación. Este se basa en una endonucleasa guiada por ARN, que constituye un sistema de defensa inmune adaptativo contra fagos, virus y plásmidos de bacterias y archea. Incorporando ADN codificante para la endonucleasa Cas9, y un ARN guía para el sitio a cortar, el sistema puede ser utilizado para generar un corte de la doble hebra en el sitio puntual de la mutación e inducir una reparación por recombinación homóloga28. Ese ARN guía puede variar según la mutación que se busca reparar.
CONCLUSIŇN
El diagnóstico para esta enfermedad está en constante avance, como puede verse en las nuevas técnicas basadas en paneles de genes estudiados por next generation sequencing. Sin embargo, consideramos que el tratamiento podría mejorar. Si bien están investigando nuevos medicamentos que funcionen como quelantes de hierro (deferoxamina), los mismos siguen basándose en reducir síntomas o buscan reducir los niveles de hierro, que es el factor ambiental predisponente.
Creemos que es necesario desarrollar una terapia correctiva,que serviría no solo para la Porfiria Cutánea Tardía sino también para la porfiria hepato-eritropoyética, cuya mutación se encuentra en el mismo gen (URO-D) pero en homocigosis.
REFERENCIAS
-
1.- Blanco A, Blanco G. Medical biochemistry. Academic Press, an imprint of Elsevier; 2017.
2.- Handler NS, Handler MZ, Stephany MP, Handler GA, Schwartz RA. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56(6):e106-e117
3.- żSabe Ud. qué son las Porfirias? [Internet]. IntraMed. Disponible en: http://www.intramed.net/contenidover.asp?contenidoID=55921 [fecha de último acceso: 08/05/2018]
4.- Porphyria cutanea tarda as the most common porphyria. - Acta dermatovenerologica Croatica: ADC. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/18093456 [fecha de último acceso: 05/06/2018]
5.- Melito VA, Rossetti MV, Parera V E, Batlle A. Porfirias poco frecuentes: Casos detectados en la población argentina. Rev. argent. dermatol. [Internet]. 2006 Dic. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1851-300X2006000300003&lng=es. [fecha de último acceso: 08/05/2018].
6.- McKusick VA, #176100 Porphyria Cutanea Tarda [Internet]. 6/2/1986. [Updated 07/09/2016]. In: O'Neill MJF, editors. OMIM . Disponible en: https://www.omim.org/entry/176100 [fecha de último acceso: 05/06/2018]
7.- Méndez M, Rossetti MV, Batlle AM del C y Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52: 417-424
8.- Sánchez MF, Navarrete J, Buchroithner C. Porfiria cutánea tarda: Revisión de la literatura a propósito de un caso. Rev. Chilena Dermatol. 2015; 31(3): 265-271. Disponible en: https://www.sochiderm.org/web/revista/31_3/7.pdf [fecha de último acceso: 08/05/2018]
9.- Bocchini CA. * 613521 Uroporphyrinogen decarboxylase;[Internet]. 8/6/2010 [Updated 09/11/2013]. In: Al-Aama J, editors.OMIM. Disponible en: http://omim.org/entry/613521 [fecha de último acceso: 05/06/2018].
10.- UROD gene - Genetics Home Reference [Internet]. U.S. National Library of Medicine. National Institutes of Health; Disponible en: https://ghr.nlm.nih.gov/gene/UROD#conditions [fecha de último acceso: 05/06/2018]
11.- Ramanujam V-MS, Anderson KE. Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias. Current Protocols in Human Genetics. 2015 Jan;
12.- Pilot Trial of Deferasirox in the Treatment of Porphyria Cutanea Tarda - Full Text View [Internet]. Home - ClinicalTrials.gov. Disponible en: https://www.clinicaltrials.gov/ct2/show/NCT00599326 [fecha de último acceso: 05/06/2018]
13.- Iribas J L, Weidmann J, Reyes M A, Korol V, Coronica M, Sixto M et al . Porfiria cutánea tardía: Reporte de 5 casos. Rev. argent. dermatol. [Internet]. 2008; 89( 1 ): 45-52. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1851-300X2008000100007&lng=es. [fecha de último acceso: 05/06/2018]
14.- Batlle AM; Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. Actualizaciones Médico Bioquímicas. La Plata: Federación Bioquímica de la Provincia de Buenos Aires; 1997.
15.- Rodwell VW. Harper: bioqui?mica ilustrada. McGraw-Hill; 2015.
16.- Biolabs, New England. "Long Range PCR." New England Biolabs: Reagents for the Life Sciences Industry, www.neb.com/applications/dna-amplification-pcr-and-qpcr/specialty-pcr/long-range-pcr
17.- Liu LU, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Familial Porphyria Cutanea Tarda. 2013 Jun 6 [Updated 2016 Sep 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK143129/ [fecha de último acceso: 05/06/2018]
18.- Wallace SE. Educational Materials - Genetic Testing: Current Approaches [Internet]. GeneReviews® [Internet]. U.S. National Library of Medicine; 2018. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK279899/#app5.Multigene_Panels [fecha de último acceso: 05/06/2018]
19.- Porphyria Cutanea Tarda Type II/Hepatoerythropoietic Porphyria via UROD Gene Sequencing with CNV Detection - Tests - GTR - NCBI Advances in pediatrics. Disponible en: https://www.ncbi.nlm.nih.gov/gtr/tests/519417/methodology/ [fecha de último acceso: 05/06/2018]
20.- Chronic/Cutaneous Porphyria Sequencing Panel with CNV Detection - Tests - GTR - NCBI. Advances in pediatrics. Disponible en: https://www.ncbi.nlm.nih.gov/gtr/tests/552988/ [fecha de último acceso: 05/06/2018]
21.- CENTRO DE INVESTIGACIONES SOBRE PORFIRINAS Y PORFIRIAS [Internet]. Google Sites. Disponible en:https://sites.google.com/site/cipypconicet/Home[fecha de último acceso: 08/05/2018].
22.- Porfiria Cutánea Tarda (PCT) (internet) | European Porphyria Network Disponible en: http://porphyria.eu/es/content/porfiria-cutanea-tarda-pct#6[fecha de último acceso: 08/05/2018].
23.- Bonkovsky HL. Porfiria cutánea tardía - Trastornos endocrinos y metabólicos - Manual MSD versión para profesionales. MSD Manual Consumer Version. Disponible en: https://www.msdmanuals.com/es-ar/professional/trastornos-endocrinos-y-metab%C3%B3licos/porfirias/porfiria-cut%C3%A1nea-tard%C3%ADa#v8380733_es [fecha de último acceso: 05/06/2018]
24.- Motswaledi M. The diagnosis and management of porphyria cutanea tarda (PCT). South African Family Practice. 2009;51(3):186-187.
25.- Pandya, A. Pilot Trial of Deferasirox in the Treatment of Porphyria Cutanea Tarda (internet) - ClinicalTrials.gov Disponible en: https://www.clinicaltrials.gov/ct2/show/NCT00599326?cond=Porphyria+Cutanea+Tarda&rank=1[fecha de último acceso: 05/06/2018]
26.- Conlon TJ, Cossette T, Erger K, Choi Y-K, Clarke T, Scott-Jorgensen M, et al. Efficient Hepatic Delivery and Expression from a Recombinant Adeno-associated Virus 8 Pseudotyped ?1-Antitrypsin Vector. Molecular Therapy. 2005;12(5):867-875.
27.- Zern MA, Kresina TF. Hepatic drug delivery and gene therapy. Hepatology. 1997;25(2):484-491
28.- Aravalli RN, Belcher JD, Steer CJ. Liver-targeted gene therapy: Approaches and challenges. Liver Transplantation. 2015;21(6):718-737.
CORRESPONDENCIA:
Ornella Agnese
Instituto Universitario
Hospital Italiano de Buenos Aires,
Argentina
Email: ornella.agnese @ hospitalitaliano.org.ar